10-Q: Quarterly report pursuant to Section 13 or 15(d)
Published on November 14, 2024
Table of Contents
|
|
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
(State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) |
Title of each class |
Trading Symbol(s) |
Name of each exchange on which registered |
||
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
|
Non-accelerated filer |
☒ | Smaller reporting company | ||||
| Emerging growth company | ||||||
Table of Contents
BENITEC BIOPHARMA INC.
INDEX TO FORM 10-Q
| SPECIAL NOTE REGARDING FORWARD LOOKING STATEMENTS | 1 | |||||
| PART I-FINANCIAL INFORMATION | 2 | |||||
| ITEM 1. | 2 | |||||
| Consolidated Balance Sheets as of September 30, 2024 (Unaudited) and June 30, 2024 |
2 | |||||
| 3 | ||||||
| 4 | ||||||
| 5 | ||||||
| 6 | ||||||
| ITEM 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
21 | ||||
| ITEM 3. | 52 | |||||
| ITEM 4. | 52 | |||||
| PART II-OTHER INFORMATION | ||||||
| ITEM 1. | 53 | |||||
| ITEM 1A. | 53 | |||||
| ITEM 2. | 53 | |||||
| ITEM 3. | 53 | |||||
| ITEM 4. | 53 | |||||
| ITEM 5. | 53 | |||||
| ITEM 6. | 54 | |||||
| SIGNATURES | 55 | |||||
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Report contains forward-looking statements that are subject to a number of risks and uncertainties, many of which are beyond our control. Our forward-looking statements relate to future events or our future performance and include, but are not limited to, statements concerning our business strategy, future commercial revenues, market growth, capital requirements, new product introductions, expansion plans and the adequacy of our funding. All statements, other than statements of historical fact included in this Report, are forward-looking statements. When used in this Report, the words “could,” “believe,” “anticipate,” “intend,” “estimate,” “expect,” “may,” “continue,” “predict,” “potential,” “project,” or the negative of these terms, and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain such identifying words. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements.
Some of the risks and uncertainties that may cause our actual results, performance or achievements to differ materially from those expressed or implied by forward-looking statements include the following:
| • | the success of our plans to develop and potentially commercialize our product candidates; |
| • | the timing of the completion of preclinical studies and clinical trials; |
| • | the timing and sufficiency of patient enrollment and dosing in any future clinical trials; |
| • | the timing of the availability of data from our clinical trials; |
| • | the timing and outcome of regulatory filings and approvals; |
| • | the development of novel AAV vectors; |
| • | our potential future out-licenses and collaborations; |
| • | the plans of licensees of our technology; |
| • | the clinical utility and potential attributes and benefits of ddRNAi and our product candidates, including the potential duration of treatment effects and the potential for a “one shot” cure; |
| • | our intellectual property position and the duration of our patent portfolio; |
| • | expenses, ongoing losses, future revenue, capital needs and needs for additional financing, and our ability to access additional financing given market conditions and other factors, including our capital structure; |
| • | the length of time over which we expect our cash and cash equivalents to be sufficient to execute on our business plan; |
| • | unanticipated delays; |
| • | further research and development and the results of clinical trials possibly being unsuccessful or insufficient to meet applicable regulatory standards or warrant continued development; |
| • | the ability to enroll sufficient numbers of subjects in clinical trials; |
| • | determinations made by the U.S. Food and Drug Administration and other governmental authorities; |
| • | regulatory developments in the United States of America; |
| • | our ability to protect and enforce our patents and other intellectual property rights; |
| • | our dependence on our relationships with our collaboration partners and other third parties; |
| • | the efficacy or safety of our products and the products of our collaboration partners; |
| • | the acceptance of our products and the products of our collaboration partners in the marketplace and market competition; |
| • | sales, marketing, manufacturing and distribution requirements; |
| • | greater than expected expenses, expenses relating to litigation or strategic activities; |
| • | the impact of, and our ability to remediate, the identified material weakness in our internal controls over financial reporting; |
| • | our ability to satisfy our capital needs through increasing revenue and obtaining additional financing; and |
| • | the impact of local, regional and national and international economic conditions and events; the as well as other risks detailed under the caption “Risk Factors” in this Report and in other reports filed with the SEC. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Report, we caution you that these statements are based on a combination of facts and important factors currently known by us and our expectations of the future, about which we cannot be certain. Such statements are based on assumptions and the actual outcome will be affected by known and unknown risks, trends, uncertainties and factors that are beyond our control or ability to predict. We have based the forward-looking statements included in this Report on information available to us on the date of this Report or on the date thereof. Except as required by law we undertake no obligation to revise or update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised to consult any additional disclosures that we may make directly to you or through reports that we, in the future, may file with the SEC, including annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K. |
All forward-looking statements included herein or in documents incorporated herein by reference are expressly qualified in their entirety by the cautionary statements contained or referred to elsewhere in this Report.
1
Table of Contents
| September 30, 2024 |
June 30, 2024 |
|||||||
| (Unaudited) | ||||||||
Assets |
||||||||
Current assets: |
||||||||
Cash and cash equivalents
|
$ | $ | ||||||
Restricted cash |
||||||||
Trade and other receivables |
||||||||
Prepaid and other assets |
||||||||
Total current assets |
||||||||
Property and equipment, net |
||||||||
Deposits |
||||||||
Prepaid and other assets |
||||||||
Right-of-use |
||||||||
Total assets |
$ | $ | ||||||
Liabilities and stockholders’ equity |
||||||||
Current liabilities: |
||||||||
Trade and other payables |
$ | $ | ||||||
Accrued employee benefits |
||||||||
Lease liabilities, current portion |
||||||||
Total current liabilities |
||||||||
Non-current accrued employee benefits |
||||||||
Total liabilities |
||||||||
Commitments and contingencies (Note 11) |
||||||||
Stockholders’ equity: |
||||||||
Common stock, $ |
||||||||
Additional paid-in capital |
||||||||
Accumulated deficit |
( |
) | ( |
) | ||||
Accumulated other comprehensive loss |
( |
) | ( |
) | ||||
Total stockholders’ equity |
||||||||
Total liabilities and stockholders’ equity |
$ | $ | ||||||
| Three Months Ended September 30, | ||||||||
| 2024 | 2023 | |||||||
Revenue: |
||||||||
Licensing revenues from customers |
$ |
|
$ |
|
||||
Total revenues |
|
|
||||||
Operating expenses: |
||||||||
Royalties and license fees |
|
( |
) | |||||
Research and development |
||||||||
General and administrative |
||||||||
Total operating expenses |
||||||||
Loss from operations |
( |
) | ( |
) | ||||
Other income (loss): |
||||||||
Foreign currency transaction gain (loss) |
( |
) | ||||||
Interest income (expense), net |
( |
) | ||||||
Other income (expense), net |
( |
) | ||||||
Total other income (loss), net |
( |
) | ||||||
Net loss |
$ | ( |
) | $ | ( |
) | ||
Other comprehensive income: |
||||||||
Unrealized foreign currency translation gain (loss) |
( |
) | ||||||
Total other comprehensive income (loss) |
( |
) | ||||||
Total comprehensive loss |
$ | ( |
) | $ | ( |
) | ||
Net loss |
$ | ( |
) | $ | ( |
) | ||
Deemed dividend |
$ |
|
$ | ( |
) | |||
Net loss attributable to common stockholders |
$ | ( |
) | $ | ( |
) | ||
Net loss per share: basic and diluted |
$ | ( |
) | $ | ( |
) | ||
Weighted average number of shares outstanding: basic and diluted |
||||||||
| Common Stock | Additional Paid-in
|
Accumulated | Accumulated Other Comprehensive |
Total Stockholders’ |
||||||||||||||||||||
| Shares | Amount | Capital | Deficit | Loss | Equity | |||||||||||||||||||
| Balance at June 30, 2023 |
$ |
|
$ |
$ |
( |
) |
$
|
( |
) |
$ |
||||||||||||||
| Issuance of common stock, pre-funded warrants, and common warrants sold for cash, net of offering costs of $ |
|
— | — | |||||||||||||||||||||
| Anti-dilution adjustment to warrants |
— | — | ( |
) | — | — | ||||||||||||||||||
| Share-based compensation |
— | — | — | — | ||||||||||||||||||||
| Foreign currency translation gain |
— | — | — | — | ||||||||||||||||||||
| Net loss |
— | — | — | ( |
) | — | ( |
) | ||||||||||||||||
| |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at September 30, 2023 |
$ |
|
$ |
$ |
( |
) | $ |
( |
) | $ |
||||||||||||||
| |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at June 30, 2024 |
$ |
|
$ |
$ |
( |
) | $ |
( |
) | $ |
||||||||||||||
| Exercise of pre-funded warrants |
— | — | — | — | — | |||||||||||||||||||
| Exercise of Series 2 warrants |
— | — | — | |||||||||||||||||||||
| Exercise of common warrants |
— | — | — | |||||||||||||||||||||
| Share-based compensation |
— | — | — | — | ||||||||||||||||||||
| Foreign currency translation loss |
— | — | — | — | ( |
) | ( |
) | ||||||||||||||||
| Net loss |
— | — | — | ( |
) | — | ( |
) | ||||||||||||||||
| |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at September 30, 2024 |
$ |
|
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||||
| |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Three Months Ended September 30, |
||||||||
| 2024 | 2023 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
||||||||
| Amortization of right-of-use |
||||||||
| Share-based compensation expense |
||||||||
| Changes in operating assets and liabilities: |
||||||||
| Trade and other receivables |
|
|||||||
| Prepaid and other assets |
||||||||
| Trade and other payables |
( |
) | ||||||
| Accrued employee benefits |
( |
) | ||||||
| Lease liabilities |
( |
) | ( |
) | ||||
| |
|
|
|
|||||
| Net cash used in operating activities |
( |
) | ( |
) | ||||
| |
|
|
|
|||||
| Cash flows from investing activities: |
||||||||
| Net cash used in investing activities |
|
|
||||||
| |
|
|
|
|||||
| Cash flows from financing activities: |
||||||||
| Proceeds from exercise of pre-funded warrants, series 2 warrants and common warrants |
||||||||
| Share issue transaction costs |
|
( |
) | |||||
| |
|
|
|
|||||
| Net cash provided by financing activities |
||||||||
| |
|
|
|
|||||
| Effects of exchange rate changes on cash, cash equivalents, and restricted cash |
( |
) | ||||||
| |
|
|
|
|||||
| Net increase in cash, cash equivalents, and restricted cash |
||||||||
| Cash, cash equivalents, and restricted cash, beginning of period |
||||||||
| |
|
|
|
|||||
| Cash, cash equivalents, and restricted cash, end of period |
$ | $ | ||||||
| |
|
|
|
|||||
| Supplemental disclosure of cash flow information |
||||||||
| Deemed dividend |
$ |
|
$ | |||||
| |
|
|
|
|
|
|
|
|
|
Principal place of
business/country of
incorporation
|
||
| Benitec Biopharma Proprietary Limited (“BBL”) | ||
| Benitec Australia Proprietary Limited | ||
| Benitec Limited | ||
| Benitec, Inc. | ||
| Benitec LLC | ||
| RNAi Therapeutics, Inc. | ||
| Tacere Therapeutics, Inc. | ||
| Benitec IP Holdings, Inc. |
| Level 1: | Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities. | |
| Level 2: | Inputs, other than quoted prices that are observable, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. | |
| Level 3: | Unobservable inputs in which little or no market data exists, therefore developed using estimates and assumptions developed by us, which reflect those that a market participant would use. | |
| Software |
|
|
| Lab equipment |
|
|
| Computer hardware |
|
|
| Leasehold improvements |
| (US$’000) | September 30, 2024 |
June 30, 2024 |
||||||
Cash at bank |
$ | $ | ||||||
Restricted cash |
||||||||
Total |
$ | $ | ||||||
| (US$’000) | September 30, 2024 |
June 30, 2024 |
||||||
Prepaid expenses |
$ | $ | ||||||
Market value of listed shares |
||||||||
Total other assets |
||||||||
Less: non-current portion |
( |
) | ( |
) | ||||
Current portion |
$ | $ | ||||||
| (US$’000) | September 30, 2024 |
June 30, 2024 |
||||||
Software |
$ | $ | ||||||
Lab equipment |
||||||||
Computer hardware |
||||||||
Leasehold improvements |
||||||||
Total property and equipment, gross |
||||||||
Accumulated depreciation and amortization |
( |
) | ( |
) | ||||
Total property and equipment, net |
$ | $ | ||||||
| (US$’000) | September 30, 2024 |
June 30, 2024 |
||||||
Trade payable |
$ | $ | ||||||
Accrued consultant fees |
||||||||
Accrued professional fees |
||||||||
Accrued clinical development project costs |
||||||||
Accrued patent expenses |
|
|||||||
Other payables |
||||||||
Total |
$ | $ | ||||||
| (US$’000) | Operating lease right- of- use assets |
|||
Balance at July 1, 2024 |
$ | |||
Amortization of right of use asset |
( |
) | ||
Operating lease right-of-use |
$ | |||
| (US$’000) | Operating lease liabilities |
|||
Balance at July 1, 2024 |
$ | |||
Principal payments on operating lease liabilities |
( |
) | ||
Operating lease liabilities at September 30, 2024 |
||||
Less: non-current portion |
|
|||
Current portion at September 30, 2024 |
$ | |||
| (US$’000) | September 30, 2024 |
|||
2025 |
$ | |||
Total operating lease payments |
||||
Less imputed interest |
( |
) | ||
Present value of operating lease liabilities |
$ | |||
| Common Stock from Warrants |
Weighted- average Exercise Price (per share) |
|||||||
Outstanding at July 1, 2024 |
$ | |||||||
Pre-funded warrants exercised |
$ | |||||||
Series 2 warrants exercised |
$ | |||||||
Common warrants exercised |
$ | |||||||
Outstanding and exercisable at September 30, 2024 |
$ | |||||||
| Stock Options |
Weighted- average Exercise Price |
Weighted- average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
Outstanding at June 30, 2024 |
$ |
|
$ | |||||||||||||
Granted |
|
|
— |
— | ||||||||||||
Expired |
|
|
— |
— | ||||||||||||
Forfeited |
|
|
— |
— | ||||||||||||
Outstanding at September 30, 2024 |
$ |
|
$ | |||||||||||||
Exercisable at September 30, 2024 |
$ |
|
$ | |||||||||||||
| Three Months Ended September 30, |
||||||||
| (US$’000) | 2024 | 2023 | ||||||
Research and development |
$ | $ | ||||||
General and administrative |
||||||||
Total share-based compensation expense |
$ | $ | ||||||
Table of Contents
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of financial condition and operating results together with our consolidated financial statements and the related notes and other financial information included elsewhere in this document.
Company Overview
We endeavor to become the leader in discovery, development, and commercialization of therapeutic agents capable of addressing significant unmet medical need via the application of the silence and replace approach to the treatment of genetic disorders.
Benitec Biopharma Inc. (“Benitec” or the “Company” or in the third person, “we” or “our”) is a clinical-stage biotechnology company focused on the advancement of novel genetic medicines with headquarters in Hayward, California. The proprietary platform, called DNA-directed RNA interference, or ddRNAi, combines RNA interference, or RNAi, with gene therapy to create medicines that facilitate sustained silencing of disease-causing genes following a single administration. The unique therapeutic constructs also enable the simultaneous delivery of wildtype replacement genes, facilitating the proprietary “silence and replace” approach to the treatment of genetically defined diseases. The Company is developing a silence and replace-based therapeutic (BB-301) for the treatment of Oculopharyngeal Muscular Dystrophy (OPMD), a chronic, life-threatening genetic disorder.
BB-301 is a silence and replace-based genetic medicine currently under development by Benitec. BB-301 is an AAV-based gene therapy designed to permanently silence the expression of the disease-causing gene (to slow, or halt, the biological mechanisms underlying disease progression in OPMD) and to simultaneously replace the mutant gene with a wildtype gene (to drive restoration of function in diseased cells). This fundamental therapeutic approach to disease management is called “silence and replace.” The silence and replace mechanism offers the potential to restore the normative physiology of diseased cells and tissues and to improve treatment outcomes for patients suffering from the chronic, and potentially fatal, effects of OPMD. BB-301 has been granted Orphan Drug Designation in the United States and the European Union.
The targeted gene silencing effects of RNAi, in conjunction with the durable transgene expression achievable via the use of modified viral vectors, imbues the silence and replace approach with the potential to produce permanent silencing of disease-causing genes along with simultaneous replacement of the wild type gene function following a single administration of the proprietary genetic medicine. We believe that this novel mechanistic profile of the current and future investigational agents developed by Benitec could facilitate the achievement of robust and durable clinical activity while greatly reducing the frequency of drug administration traditionally expected for medicines employed for the management of chronic diseases. Additionally, the achievement of permanent gene silencing and gene replacement may significantly reduce the risk of patient non-compliance during the course of medical management of potentially fatal clinical disorders.
We will require additional financing to progress our product candidates through to key inflection points.
Our proprietary technology platforms are designated as DNA-directed RNA interference, or “ddRNAi”, and “silence and replace.” ddRNAi is designed to produce permanent silencing of disease-causing genes, by combining RNA interference, or RNAi, with viral delivery agents typically associated with the field of gene therapy (i.e., viral vectors). Modified AAV vectors are employed to deliver genetic constructs which encode short hairpin RNAs that are, then, serially expressed and processed to produce siRNA molecules within the transduced cell for the duration of the life of the target cell. These newly introduced siRNA molecules drive
21
Table of Contents
permanent silencing of the expression of the disease-causing gene. The silence and replace approach further bolsters the biological benefits of permanent silencing of disease-causing genes by incorporating multifunctional genetic constructs within the modified AAV vectors to create an AAV-based gene therapy agent that is designed to silence the expression of disease-causing genes (to slow, or halt, the underlying mechanism of disease progression) and to simultaneously replace the mutant genes with normal, “wildtype” genes (to drive restoration of function in diseased cells). This fundamentally distinct therapeutic approach to disease management offers the potential to restore the underlying physiology of the treated tissues and, in the process, improve treatment outcomes for patients suffering from the chronic and, potentially, fatal effects of diseases like Oculopharyngeal Muscular Dystrophy (OPMD).
Traditional gene therapy is defined by the introduction of an engineered transgene to correct the pathophysiological derangements derived from mutated or malfunctioning genes. Mutated genes can facilitate the intracellular production of disease-causing proteins or hamper the production of critical, life-sustaining, proteins. The introduction of a new transgene can facilitate the restoration of production of normal proteins within the diseased cell, thus restoring natural biological function. Critically, the implementation of this traditional method of gene therapy cannot eliminate the expression, or the potential deleterious effects of, the underlying mutant gene (as mutant proteins may be continually expressed and aggregate or drive the aggregation of other native proteins within the diseased cell). In this regard, the dual capabilities of the proprietary silence and replace approach to silence a disease-causing gene via ddRNAi and simultaneously replace the wild type activity of a mutant gene via the delivery of an engineered transgene could facilitate the development of differentially efficacious treatments for a range of genetic disorders.
Overview of RNAi and the siRNA Approach
The mutation of a single gene can cause a chronic disease via the resulting intracellular production of a disease-causing protein (i.e., an abnormal form of the protein of interest), and many chronic and/or fatal disorders are known to result from the inappropriate expression of a single gene or multiple genes. In some cases, genetic disorders of this type can be treated exclusively by “silencing” the intracellular production of the disease-causing protein through well-validated biological approaches like RNA interference (“RNAi”). RNAi employs small nucleic acid molecules to activate an intracellular enzyme complex, and this biological pathway temporarily reduces the production of the disease-causing protein. In the absence of the disease-causing protein, normal cellular function is restored and the chronic disease that initially resulted from the presence of the mutant protein is partially or completely resolved. RNAi is potentially applicable to over 20,000 human genes and a large number of disease-causing microorganism-specific genes.
22
Table of Contents
Figure 1
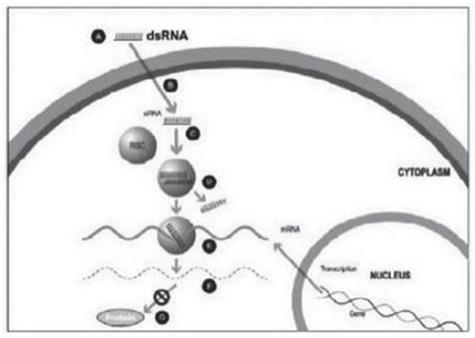
A small double stranded RNA, or dsRNA, molecule (A, Figure 1), comprising one strand known as the sense strand and another strand known as the antisense strand, which are complementary to each other, is synthesized in the laboratory. These small dsRNAs are called small interfering RNAs, or siRNAs. The sequence of the sense strand corresponds to a short region of the target gene mRNA. The siRNA is delivered to the target cell (B, Figure 1), where a group of enzymes, referred to as the RNA-Induced Silencing Complex, or RISC, process the siRNA (C, Figure 1), where one of the strands (usually the sense strand) is released (D, Figure 1). RISC uses the antisense strand to find the mRNA that has a complementary sequence (E, Figure 1) leading to the cleavage of the target mRNA (F, Figure 1). As a consequence, the output of the mRNA (protein production) does not occur (G, Figure 1). Several companies, including Alnylam Pharmaceuticals Inc. (“Alnylam”), utilize this approach in their RNAi product candidates.
Importantly, many genetic disorders are not amenable to the traditional gene silencing approach outlined in Figure 1, as the diseased cells may produce a mixture of the wild type protein of interest and the disease-causing mutant variant of the protein, and the underlying genetic mutation may be too small to allow for selective targeting of the disease-causing variant of the protein through the use of siRNA-based approaches exclusively. In these cases, it is extraordinarily difficult to selectively silence the disease-causing protein without simultaneously silencing the wild type intracellular protein of interest whose presence is vital to the conduct of normal cellular functions.
Our proprietary silence and replace technology utilizes the unique specificity and robust gene silencing capabilities of RNAi while overcoming many of the key limitations of siRNA-based approaches to disease management.
In the standard RNAi approach, double-stranded siRNA is produced synthetically and, subsequently, introduced into the target cell via chemical modification of the RNA or alternative methods of delivery. While efficacy has been demonstrated in several clinical indications through the use of this approach, siRNA-based approaches maintain a number of limitations, including:
| • | Clinical management requires repeat administration of the siRNA-based therapeutic agent for multiple cycles to maintain efficacy; |
23
Table of Contents
| • | Long-term patient compliance challenges due to dosing frequencies and treatment durations; |
| • | Therapeutic concentrations of siRNA are not stably maintained because the levels of synthetic siRNA in the target cells decrease over time; |
| • | Novel chemical modifications or novel delivery materials are typically required to introduce the siRNA into the target cells, making it complicated to develop a broad range of therapeutics agents; |
| • | Potential adverse immune responses, resulting in serious adverse effects; |
| • | Requirement for specialized delivery formulations for genetic disorders caused by mutations of multiple genes; and |
| • | siRNA acts only to silence genes and cannot be used to replace defective genes with normally functioning genes. |
Our Approach to the Treatment of Genetic Diseases—ddRNAi and Silence and Replace
Our proprietary silence and replace approach to the treatment of genetic diseases combines RNAi with wild type gene replacement to drive permanent silencing of disease-causing genes and concomitant restoration of functional wild type genes following a single administration of the therapeutic agent. Benitec employs ddRNAi in combination with classical gene therapy (i.e., transgene delivery via viral vectors) to overcome several of the fundamental limitations of RNAi.
The silence and replace approach to the treatment of genetic disorders employs adeno-associated viral vectors (“AAVs”) to deliver genetic constructs which may, after a single administration to the target tissues:
| • | Chronically express RNAi molecules inside of the target, diseased, cells (to serially silence the intracellular production of mutant, disease-causing, protein and the wild type protein of interest); |
| • | Simultaneously drive the expression of a wild type variant of the protein of interest (to restore native intracellular biological processes); and |
| • | AAV vectors can accommodate the multi-functional DNA expression cassettes containing the engineered wild type transgenes and the novel genes encoding short hairpinRNA/microRNA molecules (shRNA/miRNA) that are required to support the development of therapeutic agents capable of the achievement of the goals of the silence and replace approach to therapy. |
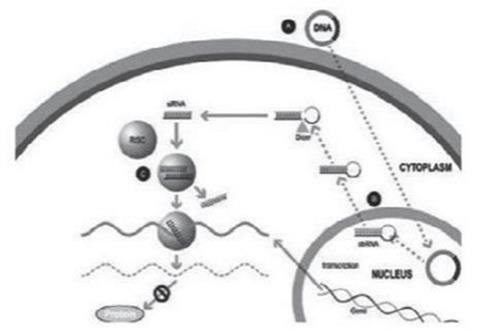
Our silence and replace technology utilizes proprietary DNA expression cassettes to foster continuous production of gene silencing shRNAs and wild type proteins (via expression of the wild type transgene). A range of viral gene therapy vectors can be used to deliver the DNA construct into the nucleus of the target cell and, upon delivery, shRNA molecules are expressed and subsequently processed by intracellular enzymes into siRNA molecules that silence the expression of the mutant, disease-causing protein (Figure 2).
In the silence and replace approach (Figure 2):
| • | A DNA construct is delivered to the nucleus of the target cell by a gene therapy vector (A) such as an AAV vector; |
| • | Once inside of the nucleus, the DNA construct drives the continuous production of shRNA molecules (B) which are processed by an enzyme called Dicer into siRNAs (C); |
| • | The processed siRNA is incorporated into RISC and silences the target gene using the same mechanism shown in Figure 1; and |
| • | When the DNA expression cassette is additionally comprised of a wild type transgene, upon entry of the DNA construct into the nucleus of the target cell via the use of the AAV vector, the DNA construct also drives the continuous production of wild type protein (to restore native intracellular biological processes). |
24
Table of Contents
Figure 2
Our strategy is to discover, develop and commercialize treatments that leverage the capabilities of ddRNAi and the silence and replace approach to disease management.
For selected product candidates, at the appropriate stage, we may collaborate with large biopharmaceutical companies to further co-develop and, if approved, commercialize our ddRNAi-based and silence and replace-based products to achieve broad clinical and commercial distribution. For specific clinical indications that we deem to be outside of our immediate areas of focus, we will continue to out-license, where appropriate, applications of our ddRNAi and silence and replace technology to facilitate the development of differentiated therapeutics, which could provide further validation of our proprietary technology and approach to disease management.
Our cash and cash equivalents will be deployed for the advancement of our product candidate BB-301 for the treatment of OPMD-derived dysphagia, including the natural history lead-in study and the Phase 1b/2a BB-301 treatment study, for the continued advancement of development activities for other existing and new product candidates, for general corporate purposes and for strategic growth opportunities.
Oculopharyngeal Muscular Dystrophy—OPMD
OPMD is an insidious, autosomal-dominant, late-onset degenerative muscle disorder that typically presents in patients at 40-to-50 years of age. The disease is characterized by progressive swallowing difficulties (dysphagia) and eyelid drooping (ptosis). OPMD is caused by a specific mutation in the poly(A)-binding protein nuclear 1, or PABPN1, gene. OPMD is a rare disease; however, patients have been diagnosed with OPMD in at least 33 countries. Patient populations suffering from OPMD are well-identified, and significant geographical clustering has been noted for patients with this disorder, which could simplify clinical development and global commercialization efforts.
BB-301 is an AAV-based gene therapy designed to silence the expression of disease-causing genes (to slow, or halt, the underlying mechanism of disease progression) and to simultaneously replace the mutant genes with normal, “wildtype” genes (to drive restoration of function in diseased cells). This fundamental therapeutic approach to disease management is called “silence and replace” and this biological mechanism offers the potential to restore the underlying physiology of the treated tissues and, in the process, improve treatment outcomes for patients suffering from the chronic and, potentially, fatal effects of Oculopharyngeal Muscular Dystrophy (OPMD). BB-301 has been granted Orphan Drug Designation in the United States and the European Union.
25
Table of Contents
Our Strengths
We believe that the combination of our proprietary ddRNAi and silence and replace technology, and our deep expertise in the design and development of genetic medicines, will enable us to achieve and maintain a leading position in gene silencing and gene therapy for the treatment of human disease. Our key strengths include:
| • | A first mover advantage for silence and replace-based therapeutics; |
| • | A proprietary ddRNAi-based silence and replace technology platform that may potentially enable the serial development of single-administration therapeutics capable of facilitating sustained, long-term silencing of disease-causing genes and concomitant replacement of wild type gene function; |
| • | A proprietary AAV vector technology which improves the endosomal escape capability of virus produced in insect cells using a baculovirus system. This technology has broad application in AAV-based gene therapies; |
| • | The capabilities to drive the development of a pipeline of programs focused on chronic diseases with either large patient populations, or rare diseases, which may potentially support the receipt of Orphan Drug Designation, including OPMD; and |
| • | A growing portfolio of patents protecting improvements to our ddRNAi, and silence and replace, technology and product candidates through at least 2036, with additional patent life anticipated through at least 2040. |
Our Strategy
We endeavor to become the leader in discovery, development, and commercialization of silence and replace-based therapeutic agents. We apply the following general strategy to drive the Company towards these goals:
| • | Selectively develop proprietary and partnered programs; and |
| • | Continue to explore and secure research and development partnerships with global biopharmaceutical companies supported by the differentiated nature of our scientific platform and intellectual property portfolio. |
Our senior leadership team will continue to explore partnership opportunities with global biopharmaceutical companies, as we expect that the unique attributes of the proprietary ddRNAi and silence and replace approaches, and the breadth of potential clinical indications amenable to our proprietary methods, to support the formation of collaborations over a broad range of diseases with significant unmet medical need.
We seek to actively protect our intellectual property and proprietary technology. These efforts are central to the growth of our business and include:
| • | Seeking and maintaining patents claiming our ddRNAi and silence and replace technologies and other inventions relating to our specific products in development or that are otherwise commercially and/or strategically important to the development of our business; |
| • | Protecting and enforcing our intellectual property rights; and |
| • | Strategically licensing intellectual property from third parties to advance development of our product candidates. |
26
Table of Contents
Our Pipeline
The following table sets forth our current product candidate and the development status:
Table 1. Pipeline: Oculopharyngeal Muscular Dystrophy

We are developing BB-301 for the treatment of Oculopharyngeal Muscular Dystrophy (OPMD). The Investigational New Drug (IND) application for BB-301 was approved to proceed by the U.S. Food and Drug Administration in June 2023. The first study subject was safely treated in the BB-301 Phase 1b/2a clinical trial (NCT06185673) in November 2023. The second study subject was safely treated in February 2024, and the third study subject was safely treated in October 2024. BB-301 is the lead investigational gene therapy agent under development by Benitec, and the key attributes of BB-301 are outlined in Figure 3.
27
Table of Contents
Figure 3

28
Table of Contents
BB-301 is a first-in-class genetic medicine employing the “silence and replace” approach for the treatment of OPMD. OPMD is an insidious, autosomal-dominant, late-onset, degenerative muscle disorder that typically presents in patients at 40-to-50 years of age. The disease is characterized by progressive swallowing difficulties (dysphagia) and eyelid drooping (ptosis). OPMD is caused by a specific mutation in the poly(A)-binding protein nuclear 1 gene (PABPN1).
OPMD is a rare disease, however, patients have been diagnosed with OPMD in at least 33 countries. Patient populations suffering from OPMD are well-identified, and significant geographical clustering has been noted for patients with this disorder. Each of these attributes could facilitate efficient clinical development and global commercialization of BB-301.
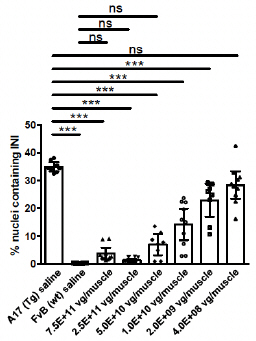
PABPN1 is a ubiquitous factor that promotes the interaction between the poly(A) polymerase and CPSF (cleavage and polyadenylation specificity factor) and, thus, controls the length of mRNA poly(A) tails, mRNA export from the nucleus, and alternative poly(A) site usage. The characteristic genetic mutation underlying OPMD results in trinucleotide repeat expansion(s) within exon 1 of PABPN1 and results in an expanded poly-alanine tract at the N-terminal end of PABPN1. The mutation generates a protein with an N-terminal expanded poly-alanine tract of up to 18 contiguous alanine residues, and the mutant protein is prone to the formation of intranuclear aggregates designated as intranuclear inclusions (INIs). The INIs that sequester wildtype PABPN1 may contribute to the “loss of function” phenotype associated with OPMD.
No therapeutic agents are approved for the treatment of OPMD. Additionally, there are no surgical interventions available to OPMD patients that modify the natural history of the disease, which is principally comprised of chronic deterioration of swallowing function. BB-301 has received Orphan Drug Designation in the United States and the European Union and, upon achievement of regulatory approval for BB-301 in these respective jurisdictions, the Orphan Drug Designations would provide commercial exclusivity independent of intellectual property protection. While OPMD is a rare medical disorder, we believe the commercial opportunity for a safe and efficacious therapeutic agent in this clinical indication exceeds $1 billion over the course of the commercial life of the product.
Investigational therapies that have been explored, unsuccessfully, in the past include:
| • | Intravenous administration of trehalose; and |
| • | The use of autologous myoblast transplant. |
BB-301 is our Lead, Silence and Replace-Based, OPMD Therapeutic Agent
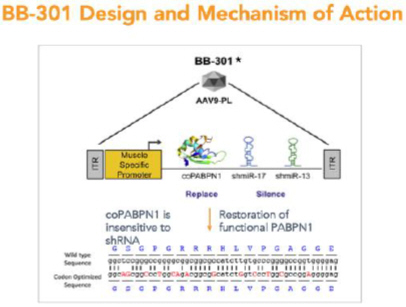
BB-301 is composed of a modified AAV serotype 9 (AAV9) capsid that expresses a bifunctional construct under the control of a single muscle specific Spc5-12 promoter to achieve co-expression of both the codon-optimized PABPN1 mRNA and two shmiR molecules directed against wild type and mutant PABPN1. BB-301 is designed to correct the genetic defect underlying OPMD following a single localized administration.
BB-301—Design and Mechanism of Action
BB-301 is designed to target two distinct regions of the PABPN1 mRNA to accomplish gene silencing via the concomitant expression of two distinct shmiRs from a single DNA construct (Figure 4). BB-301 is also engineered to drive the simultaneous expression of a codon-optimized, siRNA-resistant, version of the wild type PABPN1 gene (Figure 4).
29
Table of Contents
Figure 4
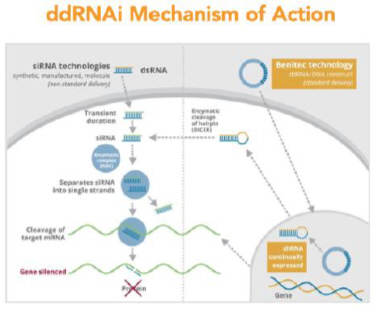
In collaboration with researchers at the Royal Holloway University of London and the Institut de Myologie in Paris, we developed a ddRNAi construct expressing three shRNAs against three distinct regions of PABPN1 mRNA and observed effective silencing of the PABPN1 gene in vitro using this ddRNAi construct. Furthermore, as part of this collaboration, we have generated a gene expression construct that produces a siRNA- resistant version of the wild type PABPN1 gene.
In subsequent studies undertaken exclusively by Benitec, a second set of target regions within PABPN1 were identified for therapeutic development and shmiRs designed against these regions. Additional shmiRs have also been designed for the original shRNA developed in collaboration with Royal Holloway University of London and the Institut de Myologie. The ‘silence and replace’ construct, designated BB-301, incorporates the two best performing shmiRs, and the gene expression construct that produces a siRNA-resistant version of the wild type PABPN1 gene, under the control of a muscle-specific promoter. The mechanism of action of BB-301 is shown in Figure 5.
Figure 5
30
Table of Contents
In initial in vivo studies evaluating the use of direct intramuscular injection of AAV-based constructs with the potential to facilitate the desired silence and replace approach in the A17 transgenic mouse model of OPMD at the Royal Holloway University of London and the Institut de Myologie, we observed decreases in muscle fibrosis, increases in cross sectional area of the treated muscles, decreases in intranuclear inclusions, and normalization of muscle strength. These nonclinical results were published in Nature Communications in April 2017.
In subsequent studies, Benitec demonstrated in a key nonclinical model (the A17 mouse model) that a single intramuscular injection of BB-301 results in robust intracellular silencing of PABPN1 protein production and concomitant expression of the normal, biologically functional PABPN1 protein. In the A17 mouse model, the treatment restores muscle strength and muscle weight to wild type levels and improves other physiological hallmarks of the disease (Figure 6a, Figure 6b, Figure 6c, Figure 6d):
| • | Multiple A17 animal cohorts received single doses of BB-301 (over a range of doses spanning 4x108 vg/muscle-to-7.5x1011 vg/muscle) and, following BB-301 administration, each cohort was observed for 14-weeks |
| • | BB-301 was injected into the Tibialis Anterior (TA) muscle of 10 week old-to-12 week old animals and, 14-weeks post administration, each A17 cohort was anesthetized and the contractile properties of the injected TA muscles were analyzed via in-situ muscle electrophysiology |
| • | Intermediate doses of BB-301 resulted in 75% silencing of PABPN1 and 26% replacement of wild type PABPN1 activity, leading to full restoration of muscle strength, clearance of INIs, and a reduction of fibrosis |
| • | An additional experiment conducted over the course of 20-weeks demonstrated that more modest doses of BB-301 (which supported only partial resolution of the disease phenotype at week-14) were, surprisingly, able to facilitate significant benefit at 20-weeks, as evidenced by restoration of parameters relating to muscle strength, weight and INI formation |
Figure 6a. Dose-Dependent shRNA Expression
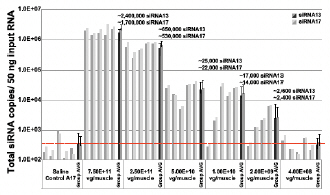
31
Table of Contents
Figure 6b. Dose-Dependent PABPN1 Inhibition and Transgene Expression
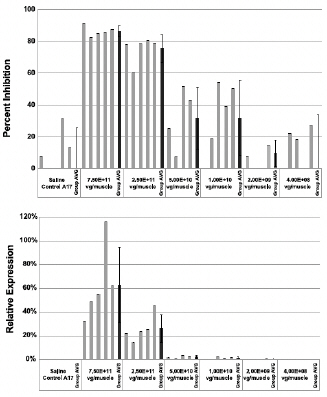
Figure 6c. Dose-Dependent Decreases in Intranuclear Inclusions
32
Table of Contents
Figure 6d. Dose-Dependent Increases in Muscle Force
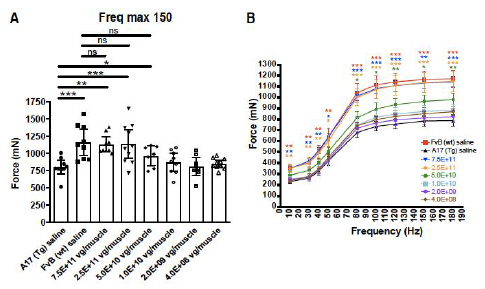
Restoration of muscle strength was assessed by muscle contractility measurements in response to a series of induced impulses that ranged from 10 to 180 Hz
Ongoing Development Activities for BB-301
On July 8, 2020, Benitec announced the initiation of the BB-301 Pilot Dosing Study in large animal subjects.
The BB-301 Pilot Dosing Study was the first of two planned CTA-enabling and IND-enabling studies that were designed to be conducted in large animals. The BB-301 Pilot Dosing Study was carried out under the guidance of the scientific team at Benitec, with key elements of the study design and execution conducted in close collaboration with a team of leading experts in both medicine and surgery that have been deeply engaged in the treatment of OPMD patients for several decades. The BB-301 Pilot Dosing Study, along with the subsequent GLP Toxicology and Biodistribution Study, were conducted in canine subjects and were carried out to support the validation and optimization of the newly designed method of BB-301 administration, confirm the efficiency of vector transduction and transgene expression in the key tissue compartments underlying the natural history of OPMD, confirm the optimal drug doses in advance of initiation of human clinical studies, and facilitate observation of key toxicological data-points.
The BB-301 Pilot Dosing Study was designed as an 8-week study in Beagle dogs to confirm the transduction efficiency of BB-301 upon administration via direct intramuscular injection into specific anatomical regions of the pharynx through the use of an open surgical procedure. This new route of BB-301 administration was developed in collaboration with key surgical experts in the field of Otolaryngology, and this novel method of BB-301 dosing was implemented to significantly enhance the ability of a treating physician to accurately administer the AAV-based investigational agent to the muscles that underlie the characteristic deficits associated with the progression of OPMD. It is important to note that prior nonclinical studies of BB-301 have reproducibly validated the robust biological activity achieved following direct intramuscular injection. As an example, direct injection of BB-301 into the tibialis anterior muscles of A17 mice facilitated robust transduction of the targeted skeletal muscle cells and supported complete remission of the OPMD disease phenotype in this animal model.
Benitec conducted the BB-301 Pilot Dosing Study in Beagle dog subjects to demonstrate that direct intramuscular injection of BB-301 via the use of a proprietary dosing device in an open surgical procedure could safely achieve the following goals:
| • | Biologically significant and dose-dependent levels of BB-301 tissue transduction (i.e., delivery of the multi-functional BB-301 genetic construct into the target pharyngeal muscle cells); |
33
Table of Contents
| • | Broad-based and dose-dependent expression of the three distinct genes comprising the BB-301 gene construct within the pharyngeal muscle cells; and |
| • | Durable and biologically significant levels of target gene knock-down (i.e., inhibition of the expression of the gene of interest) within the pharyngeal muscle cells. |
The Pilot Dosing Study evaluated the safety and biological activity of two concentrations of BB-301 (1.0+E13 vg/mL and 3.0+E13 vg/mL) across three distinct doses (1.0+E13 vg/mL, 3.0+E13 vg/mL with a low injection volume, and 3.0+E13 vg/mL with a high injection volume) following direct intramuscular injection into the Hypopharyngeal (HP) muscles and the Thyropharyngeal (TP) muscles of Beagle dogs via the use of a proprietary delivery device employed in an open surgical procedure. The HP muscle in Beagle dogs corresponds to the Middle Pharyngeal Constrictor muscle in human subjects, and the TP muscle in Beagle dogs corresponds to the Inferior Pharyngeal Constrictor muscle in human subjects. Atrophy, fibrosis, and the presence of intranuclear inclusions characterize the Middle Pharyngeal Constrictor muscles and the Inferior Pharyngeal Constrictor muscles of human subjects diagnosed with OPMD. BB-301 was injected into the pharyngeal muscles of the Beagle dog subjects only on Day 1 of the Pilot Dosing Study, and the corresponding canine pharyngeal muscles were harvested for analysis after 8 weeks of observation post-dosing. BB-301 dosing was carried out independently by both a veterinary surgeon and a practicing Otolaryngologist who has extensive experience with the provision of palliative surgical care for OPMD patients.
The key results are summarized here:
Figure 7. Pharyngeal Muscle Tissue Transduction Levels for BB-301
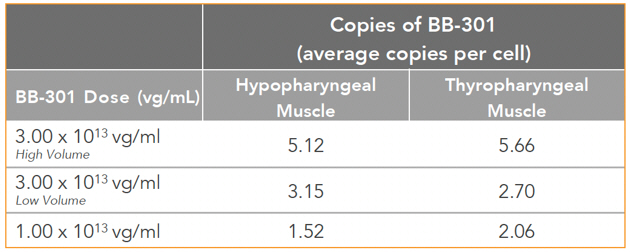
Regarding Gene Expression Levels Observed for BB-301 Within the Pharyngeal Muscle Tissues (Figure 8, Figure 9):
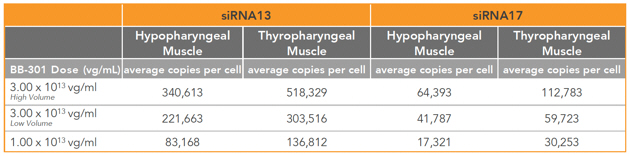
| • | BB-301 encodes two distinct siRNA species (i.e., siRNA13 and siRNA17) which are each, independently, capable of inhibiting (i.e., “silencing”) the expression of the mutant form of the PABPN1 protein and the wild type (i.e., endogenous) form of the PABPN1 protein (importantly, the mutant form of the PABPN1 protein underlies the development and progression of OPMD). |
34
Table of Contents
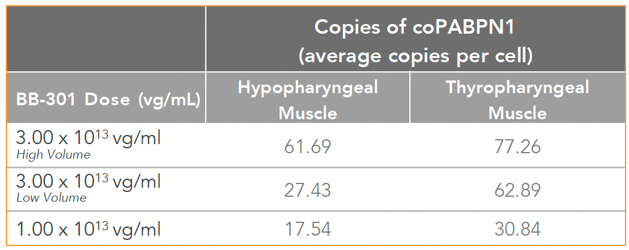
| • | BB-301 also codes for a wild type version of the PABPN1 protein whose intracellular expression is unaffected by the inhibitory activities of siRNA13 and siRNA17, and this codon optimized PABPN1 protein (i.e., coPABPN1) serves to replenish the endogenous form of the PABPN1 protein and to replace the mutant form of PABPN1 that underlies the development and progression of OPMD in diseased tissues. |
| • | For comparative purposes, it should be noted that the average range of expression for wild type PABPN1 within the pharyngeal muscle cells of Beagle dogs is 4.5 copies per cell-to-7.8 copies per cell. |
Figure 8. siRNA13 Expression Levels for BB-301 within Pharyngeal Muscle Tissues and siRNA17 Expression Levels for BB-301 within Pharyngeal Muscle Tissues
35
Table of Contents
Figure 9. coPABPN1 Expression Levels for BB-301 within Pharyngeal Muscle Tissues
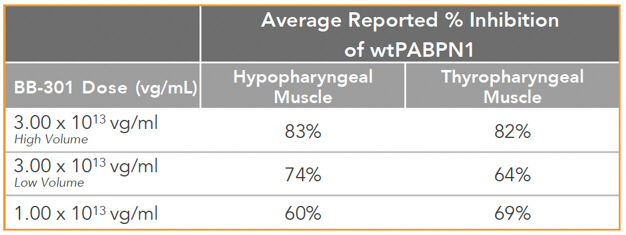
Regarding Wild Type PABPN1 Silencing (i.e. target “knock-down”) Observed for BB-301 Within the Pharyngeal Muscle Tissues (Figure 10):
| • | As noted above, BB-301 encodes two distinct siRNA species (i.e., siRNA13 and siRNA17) which are each, independently, capable of inhibiting (i.e., “silencing”) the expression of all forms of the PABPN1 protein (siRNA13 and siRNA17 silence the expression of both wild type PABPN1 [wtPABPN1] and mutant PABPN1). |
| • | While the Beagle dog subjects treated in the BB-301 Pilot Dosing Study did not express mutant PABPN1, the level of BB-301-driven gene silencing for the PABPN1 target can be accurately assessed due to the equivalent inhibitory effects of siRNA13 and siRNA17 on both wtPABPN1 and mutant PABPN1. |
| • | Thus, the wtPABPN1 silencing activity observed in the BB-301 Pilot Dosing Study served as a surrogate for the activity that would be anticipated in the presence of mutant PABPN1. |
| • | BB-301 has been evaluated in prior nonclinical studies in animals that express mutant PABPN1 and manifest the key signs and symptoms of OPMD and, in these animal models of OPMD, the achievement of PABPN1 silencing levels of 31% inhibition or higher led to resolution of OPMD disease symptoms and correction of the histological hallmarks of OPMD. |
36
Table of Contents
Figure 10. PABPN1 Silencing (i.e., “target knock-down”) within Pharyngeal Muscle Tissues
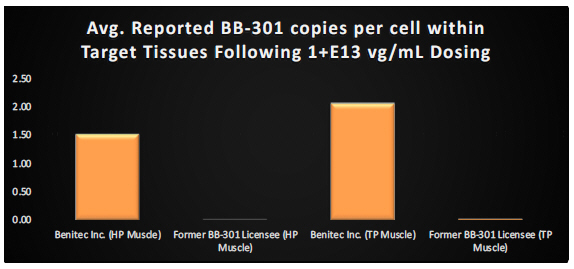
Finally, it is critical to highlight the key methodological distinctions between the BB-301 Pilot Dosing Study in Beagle dogs conducted by Benitec (i.e., the study described above) and the prior Beagle dog dosing study carried out independently by the previous BB-301 licensee. The BB-301 dosing study conducted by the prior BB-301 licensee employed non-ideal routes and methods of BB-301 administration to the target pharyngeal muscle tissues and employed similarly limited analytical methods at the completion of the dosing phase of the study. The Benitec team worked to optimize the route and method of administration of BB-301 and to refine the core analytical methods employed following the completion of dosing.
Following the implementation of these methodological modifications, Benitec demonstrated a 248-fold improvement (+24,650%) in BB-301 transduction of the HP muscle and a 111-fold improvement (+11,027%) in BB-301 transduction of the TP muscle relative to the levels of BB-301 transduction observed by the previous BB-301 licensee (Figure 11).
37
Table of Contents
Figure 11. Impact of Benitec-Initiated Methodological Improvements on the Relative Pharyngeal Muscle Tissue Transduction Levels Achieved for BB-301
Following the disclosure of the positive interim BB-301 Pilot Dosing Study results, Benitec completed pre-CTA and pre-IND meetings with regulatory agencies in France, Canada, and the United States.
Summary of the Key Regulatory Interactions:
| • | In June 2023 the U.S. Food and Drug Administration (FDA) cleared the Investigational New Drug (IND) application for BB-301 which allowed dosing of BB-301 to begin for OPMD subjects that are eligible for enrollment into the Phase 1b/2a treatment study (NCT06185673) described below. |
Operational Updates
The key milestones related to the development of BB-301 for the treatment of OPMD, along with other corporate updates, are outlined below:
BB-301 Clinical Development Program Overview:
| • | The BB-301 clinical development program will be conducted in the United States, and the primary elements of the program are summarized below: |
| • | The program will comprise approximately 76 weeks of follow-up which we anticipate will consist of: |
| • | The OPMD Natural History (NH) Study: 6-month pre-treatment observation periods for the evaluation of baseline disposition and natural history of OPMD-derived dysphagia (swallowing impairment) in each study participant. |
| • | Dosing with BB-301: 1-day of BB-301 dosing to initiate participation in the Phase 1b/2a single-arm, open-label, sequential, dose-escalation cohort study (NCT06185673). BB-301 will be delivered directly to the pharyngeal muscles of each study subject. |
| • | Phase 1b/2a Treatment Evaluation: 52-weeks of post-dosing follow-up for conclusive evaluation of the primary and secondary endpoints of the BB-301 Phase 1b/2a treatment study (NCT06185673), with interim safety and efficacy results expected to be available at the end of each 180-day period following the administration of BB-301. |
38
Table of Contents
| • | The OPMD NH Study will characterize the level of dysphagia borne by each OPMD subject at baseline and assess subsequent progression of dysphagia via the use of the following quantitative radiographic measures (i.e., videofluoroscopic swallowing studies or “VFSS”). The VFSS outlined below collectively provide objective assessments of global swallowing function and the function of the pharyngeal constrictor muscles (i.e., the muscles whose functional deterioration drives disease progression in OPMD): |
| • | Total Pharyngeal Residue %(C2-4)2 |
| • | Pharyngeal Area at Maximum Constriction (PhAMPC) |
| • | Dynamic Imaging Grade of Swallowing Toxicity Scale (DIGEST) |
| • | Vallecular Residue %(C2-4)2, Pyriform Sinus Residue %(C2-4)2, and Other Pharyngeal Residue %(C2-4)2 |
| • | Normalized Residue Ratio Scale (NRRSv, NRRSp) |
| • | Pharyngeal Construction Ratio (PCR) |
| • | The NH study will also employ clinical measures of global swallowing capacity and oral-pharyngeal dysphagia, along with two distinct patient-reported outcome instruments targeting the assessment of oral-pharyngeal dysphagia. |
| • | Upon the achievement of 6-months of follow-up in the NH Study, participants will, potentially, be eligible for enrollment into the BB-301 Phase 1b/2a treatment study (NCT06185673). |
| • | BB-301 Phase 1b/2a Treatment Study (NCT06185673): |
| • | This first-in-human (FIH) study will evaluate the safety and clinical activity of intramuscular doses of BB-301 administered to subjects with OPMD. |
| • | The primary endpoint of the FIH study will be safety. |
| • | Secondary endpoints are designed to determine the impact of BB-301 on swallowing efficiency, swallowing safety, and pharyngeal constrictor muscle function in subjects diagnosed with OPMD with dysphagia via the use of serial clinical and videofluoroscopic assessments. Critically, each of the clinical and videofluoroscopic assessments employed in the FIH study will be equivalent to those employed for the NH study to facilitate comparative clinical and statistical analyses for each study subject. |
| • | The primary and secondary endpoints will be evaluated during each 90-day period following BB-301 intramuscular injection (Day 1). |
| • | The NH of dysphagia observed for each OPMD NH Study participant, as characterized by the VFSS and clinical swallowing assessments carried out during the NH Study, will serve as the baseline for comparative assessments of safety and efficacy of BB-301 upon rollover from the NH Study onto the BB-301 Phase 1b/2a Treatment Study (NCT06185673). |
| • | In December 2022, Benitec began screening OPMD subjects for the NH Study at the lead clinical study site in the United States. |
| • | In January 2023, Benitec announced the enrollment of the first OPMD subject into the NH Study in the United States. |
| • | In November 2023, Benitec announced the completion of the administration of BB-301 to the first study subject in the Phase 1b/2a clinical study (NCT06185673) in the United States. The second study subject was treated with BB-301 in February 2024, and the third study subject was treated in October 2024. |
| • | As of January 2024, 23 subjects had enrolled into the NH study in the United States. |
| • | On October 14, 2024 Benitec reported positive interim clinical trial data for study subject 1 (9-months post-BB-301 treatment) and study subject 2 (6-months post-BB-301 treatment) in the BB-301 Phase 1b/2a Treatment Study (NCT06185673) |
Summary of Results:
| • | Two subjects have received the lowest-dose of gene therapy BB-301 (1.2e13 vg/subject), and there were no Significant Adverse Events. |
| • | Dysphagic symptoms at baseline for Subject 1 (7-years post diagnosis) were more severe than those of Subject 2 (6-years post diagnosis) as assessed by pre-dose Sydney Swallow Questionnaire (SSQ) and Total Pharyngeal Residue (TPR) results, but both Subjects experienced significant levels of clinical benefit per the post-dose SSQ scores and TPR results. |
| • | The SSQ Total Scores and SSQ Sub-Scores correlate strongly with the VFSS TPR results. |
39
Table of Contents
| • | Subject 1 experienced clinically meaningful improvements in post-dose SSQ Total Score and SSQ Sub-Scores at Day 270 driven by corresponding reductions in VFSS TPR values. |
| • | Subject 2 experienced clinically meaningful improvements in post-dose SSQ Total Score and SSQ Sub-Scores at Day 180, with an SSQ Total Score representative of a normal swallowing profile, driven by a corresponding reduction in the frequency of pathologic low-volume sequential swallows. |
| • | These data represent the first reported successful improvements in swallow function using a novel gene therapy for OPMD. |
Subjects enrolled in the NH Study and the BB-301 Phase 1b/2a Clinical Study have been shown to be impacted by:
| • | Excessive accumulation of pharyngeal residue post-swallow |
| • | Pathologic low-volume sequential swallows (i.e., rapid contractions of the pharyngeal muscles during the consumption of low volumes of thin liquids) |
Subject 1 (270 Days post-BB-301 dose):
Global inefficiency of swallowing for solid food, thin liquid, and thick liquids drives dysphagia for Subject 1.
Subject 1 displayed continued clinically meaningful reductions (i.e., improvements) in SSQ Total Score (35% reduction) and SSQ Sub-Scores (42% reduction for Thin Liquid, 16% reduction for Solid Food, and 22% reduction for Thick Liquids). Subject 1 displayed correspondingly significant reductions (i.e., improvements) in VFSS TPR (33% reduction for Thin Liquid, 18% reduction for Solid Food, and 30% reduction for Thick Liquids) following the administration of the low-dose of BB-301 as compared to the average values recorded for Subject 1 during the pre-dose period.
Subject 2 (180 Days post-BB-301-dose):
Pathologic low-volume sequential swallowing for thin liquid drives dysphagia for Subject 2. Pathologic low-volume sequential swallows are experienced by the subject as multiple swallows and are detected during VFSS as a series of rapid contractions of the pharyngeal muscles interrupting the discrete peristaltic contraction pattern typically observed during swallows of low volumes of thin liquids.
Subject 2 displayed clinically meaningful reductions (i.e., improvements) in SSQ Total Score (89% reduction) and the SSQ Sub-Score for the necessity of repeat swallows during consumption (84% reduction) as compared to the average values recorded for Subject 2 during the pre-dose period. The average post-dose SSQ Total Score of 82 is representative of a clinically normal swallowing profile for Subject 2. Subject 2 displayed correspondingly significant reductions (i.e., improvements) in the post-dose frequency of low-volume sequential swallows as evaluated by VFSS (92% reduction) following the administration of the low-dose of BB-301 as compared to the pre-dose values recorded for Subject 2 during the pre-dose period.
All study Subjects are blinded to their SSQ Total Scores and VFSS TPR assessment results, and the Central Reader for the VFSS assessments is blinded to the SSQ Total Scores and SSQ Sub-Scores for all Study Subjects.
40
Table of Contents
Manufacturing
The manufacture of the biological products required for gene therapy is complex and difficult. We do not currently own or operate manufacturing facilities for the production of preclinical, clinical or commercial quantities of any of our product candidates. We are exploring long-term manufacturing alliances with a number of potential partners to investigate manufacturing processes in order to produce materials at reasonable scale and cost of goods to support future commercialization efforts. We do not have a long-term agreement with any third-party manufacturer, but we plan to establish such a relationship with an appropriate manufacturer to serve our long-term needs.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our contract manufacturing organizations manufacture our product candidates under cGMP conditions. cGMP is a regulatory standard for the production of pharmaceuticals that will be used in humans.
41
Table of Contents
Sales and Marketing
We have not yet established sales, marketing or product distribution operations because our product candidates are in preclinical or clinical development. If we receive marketing and commercialization approval for any of our product candidates, we intend to market the product through strategic alliances and distribution agreements with third parties. In certain cases, we may market an approved product directly worldwide or in selected geographical segments. The ultimate implementation of our strategy for realizing the financial value of our product candidates is dependent on the results of clinical trials for our product candidates, the availability of funds and the ability to negotiate acceptable commercial terms with third parties.
Competition
The biopharmaceutical industry is characterized by intense and dynamic competition to develop new technologies and proprietary therapies.
Any product candidates that we successfully develop and commercialize will have to compete with existing therapies and new therapies that may become available in the future. While we believe that our proprietary technology and scientific expertise in gene silencing using ddRNAi provide us with competitive advantages, we face potential competition from many different sources, including larger and better-funded pharmaceutical, specialty pharmaceutical and biotechnology companies, as well as from academic institutions and governmental agencies and public and private research institutions that may develop potentially competitive products or technologies. We are aware of several companies focused on developing gene therapy or gene silencing product candidates.
We are not aware of any companies developing a gene therapy or gene silencing approach for OPMD. Our product candidates, if approved, would also compete with treatments that have already been approved and accepted by the medical community, patients and third-party payers.
Many of our competitors and potential competitors, alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of treatments and the commercialization of those treatments. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and subject registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new products enter the market and advanced technologies become available. We expect any treatments that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price, the level of competition and the availability of reimbursement from government and other third party-payers.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, we expect that our therapeutic products, if approved, will be priced at a significant premium over competitive products and our ability to compete may be affected in many cases by insurers or other third-party payers seeking to encourage the use of competitive products including biosimilar or generic products.
This increasingly competitive landscape may compromise the development of our product candidates.
42
Table of Contents
Royalties, milestone payments and other license fees
We are required to pay royalties, milestone payments and other license fees in connection with our licensing of intellectual property from third parties, including as discussed below.
Foreign Currency Translation and Other Comprehensive Income (Loss)
The Company’s functional currency and reporting currency is the United States dollar. BBL’s functional currency is the Australian dollar (AUD). Assets and liabilities are translated at the exchange rate in effect at the balance sheet date. Revenues and expenses are translated at the average rate of exchange prevailing during the reporting period. Equity transactions are translated at each historical transaction date spot rate. Translation adjustments arising from the use of different exchange rates from period to period are included as a component of stockholders’ equity as “Accumulated other comprehensive loss.” Gains and losses resulting from foreign currency translation are included in the consolidated statements of operations and comprehensive loss as other comprehensive income (loss).
43
Table of Contents
April 2021 Capital Raise
On April 30, 2021, the Company announced the closing of an underwritten public offering of common stock and common stock equivalents. The Company received gross proceeds of approximately $14.3 million and net proceeds of approximately $12.7 million from the offering.
September 2022 Capital Raise
On September 15, 2022, the Company announced the closing of an underwritten public offering of common stock and common stock equivalents. The Company received gross proceeds of approximately $17.9 million and net proceeds of approximately $16.0 million from the offering.
August 2023 Capital Raise
On August 11, 2023 we closed an underwritten public offering in which we sold 875,949 shares of common stock, 15,126,226 pre-funded warrants to purchase 15,126,226 shares of common stock, and 16,002,175 common warrants to purchase up to 16,002,175 shares of common stock. The pre-funded warrants were immediately exercisable until exercised in full at an exercise price of $0.0001 per share of common stock. The common warrants were immediately exercisable at a price per share of common stock of $3.86 and expire on the fifth anniversary of such initial exercisable date. The combined purchase price for each share of common stock and accompanying common warrant was $1.93, which was allocated as $1.9299 per share of common stock and $0.0001 per common warrant. Each pre-funded warrant was sold together with one common warrant at a combined price of $1.9299, which was allocated as $1.9298 per pre-funded warrant and $0.0001 per common warrant. In addition, the Company granted the underwriter a 30-day option to purchase up to 2,331,606 additional shares of common stock and/or up to 2,331,606 additional common warrants. The underwriter partially exercised this option and purchased 458,134 additional shares of common stock and 458,134 additional common warrants. Net proceeds from the offering, including the impact of the underwriter’s partial exercise of its option and net of underwriting discounts, commissions, and other offering expenses, totaled $27.9 million.
The Company has outstanding Series 2 warrants (the “Series 2 Warrants”) which are currently exercisable into 1,733,503 shares of common stock after giving effect to the Reverse Stock Split and exercises as of September 30, 2024. The Series 2 Warrants contain an exercise price adjustment mechanism providing that certain issuances of common stock (or common stock equivalents) if made at a price lower than the existing exercise price of $11.22 of such Series 2 Warrants, would reset the exercise price to such lower price. As a result of the August 11, 2023 public offering, the exercise price of the Series 2 Warrants has been automatically reset as of the closing time of such public offering to $1.9299.
April 2024 Capital Raise
On April 22, 2024 we closed a private investment in public equity (PIPE) financing in which we sold 5,749,152 shares of common stock at a price per share of $4.80 and, in lieu of shares of common stock, pre-funded warrants to purchase up to an aggregate of 2,584,239 shares of common stock at a price per pre-funded warrant of $4.7999, to certain accredited institutional investors. The pre-funded warrants were immediately exercisable until exercised in full at an exercise price of $0.0001 per share of common stock. Gross proceeds from the financing totaled $40.0 million. Net proceeds, net of commissions and other offering expenses, totaled approximately $ 37.1 million.
ATM Agreement
On October 11, 2024, the Company into a Sales Agreement (the “Sales Agreement”) with Leerink Partners LLC (the “Agent”). Pursuant to the terms of the Sales Agreement, the Company may offer and sell shares of the Company’s common stock having an aggregate offering amount of up to $75 million from time to time through the Agent. The Agent will use its commercially reasonable efforts, as the agent and subject to the terms of the Sales Agreement, to sell the shares offered. Sales of the shares, if any, may be made in sales deemed to be an “at-the-market offering” as defined in Rule 415 under the Securities Act of 1933, as amended. The Company may also agree to sell shares to the Agent as principal for its own account on terms agreed to by the Company and the Agent. The Agent will be entitled to a commission from the Company of 3.0% of the gross proceeds from the sale of shares sold under the Sales Agreement. In addition, the Company has agreed to reimburse certain expenses incurred by the Agent in connection with the offering. Shares sold pursuant to the Sales Agreement, if any, will be sold pursuant to the Company’s shelf registration statement on Form S-3 (File No. 333-277310), that was filed with the Securities and Exchange Commission, including the related prospectus, dated March 5, 2024, as supplemented by a prospectus supplement.
Results of Operations
Revenues
The Company has not generated any revenues from the sales of products. Revenues from licensing fees are included in the revenue from customers line item on our consolidated statements of operations and comprehensive loss. Our licensing fees have been generated through the licensing of our ddRNAi technology to biopharmaceutical companies. The Company did not recognize any revenue during the three months ended September 30, 2024 and September 30, 2023.
44
Table of Contents
Royalties and License Fees
Royalties and license fees consist primarily of payments we are required to remit for royalties and other payments related to in-licensed intellectual property. Under our in-license agreements, we may pay up-front fees and milestone payments and be subject to future royalties. We cannot precisely predict the amount, if any, of royalties we will owe in the future, and if our calculations of royalty payments are incorrect, we may owe additional royalties, which could negatively affect our results of operations. As our product sales increase, we may, from time to time, disagree with our third-party collaborators as to the appropriate royalties owed, and the resolution of such disputes may be costly, may consume management’s time, and may damage our relationship with our collaborators. Furthermore, we may enter into additional license agreements in the future, which may also include royalty, milestone, and other payments.
Research and Development Expenses
Research and development expenses relate primarily to the cost of conducting clinical and preclinical trials. Preclinical and clinical development costs are a significant component of research and development expenses. The Company records accrued liabilities for estimated costs of research and development activities conducted by third-party service providers, which include the conduct of preclinical studies and clinical trials, and contract manufacturing activities. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in trade and other payables on the consolidated balance sheets and within research and development expenses on the consolidated statements of operations and comprehensive loss.
The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers. The Company makes significant judgments and estimates in determining the accrued liabilities balance at the end of each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, related benefits, travel, and equity-based compensation expense. General and administrative expenses also include facility expenses, professional fees for legal, consulting, accounting and audit services and other related costs.
We anticipate that our general and administrative expenses may increase as the Company focuses on the continued development of the clinical OPMD program. The Company also anticipates an increase in expenses relating to accounting, legal and regulatory-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums and other similar costs.
On September 13, 2023, the Compensation Committee (the “Compensation Committee”) of the Company’s Board of Directors approved increases of the base salaries of Dr. Jerel Banks, the Company’s Executive Chairman and Chief Executive Officer, and Megan Boston, the Company’s Executive Director, to $655,200 and $350,784 (Ms. Boston’s salary as noted has been converted from AUD $1.00 to USD $0.64, which was the conversion rate as of September 13, 2023) respectively, each adjustment being effective as of October 1, 2023.
45
Table of Contents
Operating Expenses
The following tables sets forth a summary of our expenses for each of the periods:
| Three Months Ended | ||||||||
| September 30, | ||||||||
| 2024 | 2023 | |||||||
| (US$ 000) | ||||||||
| Royalties and license fees |
$ | — | $ | (106 | ) | |||
| Research and development |
3,585 | 4,429 | ||||||
| General and administrative |
2,206 | 1,551 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
$ | 5,791 | $ | 5,874 | ||||
|
|
|
|
|
|||||
During the three months ended September 30, 2024 and September 30, 2023, we incurred royalties and license fees expenses of zero and $(106) thousand, respectively. The credits to expense during the three months ended September 30, 2023 relate to the reversal of accruals for license fees no longer due.
During the three months ended September 30, 2024 and September 30, 2023, respectively, we incurred $3.6 million and $4.4 million in research and development expenses, respectively. Research and development expenses relate primarily to ongoing clinical development of BB-301 for the treatment of OPMD. The year-over-year decrease for the three-month period ended September 30, 2024 reflects the timing of contract manufacturing activity and the timing of payments for the OPMD Natural History and Dosing study.
General and administrative expense totaled $2.2 million and $1.6 million for the three months ended September 30, 2024 and September 30, 2023, respectively. The increase for the three-month period ended September 30, 2024 relates primarily to an increase in share based compensation of $231 thousand and corporate costs related to the filing of an At-the-Market offering and related legal fees of $282 thousand.
46
Table of Contents
The following tables sets forth a summary of our other income (loss) for each of the periods:
| Three Months Ended | ||||||||
| September 30, | ||||||||
| 2024 | 2023 | |||||||
| (US$’000) | ||||||||
| Other Income (Loss): |
||||||||
| Foreign currency transaction gain (loss) |
$ | 93 | $ | (56 | ) | |||
| Interest income (expense), net |
604 | (6 | ) | |||||
| Other income (expense), net |
35 | (18 | ) | |||||
|
|
|
|
|
|||||
| Total other income (loss), net |
$ | 732 | $ | (80 | ) | |||
|
|
|
|
|
|||||
Other income (loss), net during the three months ended September 30, 2024 and September 30,2023, which consists of foreign currency transaction gain (loss), interest income (expense), other income (expense), totaled $732 thousand and $(80) thousand, respectively. Foreign currency transaction gains and losses reflect changes in foreign exchange rates. Net interest income for the quarter ended September 30, 2024, in comparison to the loss reported for the quarter ended September 30, 2023, reflects the increase in the Company’s cash and cash equivalent balances. Other income (expense) recognized during the three months ended September 30, 2024 relates to recognition of a tax penalty refund.
Liquidity and Capital Resources
The Company has incurred cumulative losses and negative cash flows from operations since our predecessor’s inception in 1995. The Company had accumulated losses of $195 million as of September 30, 2024. We expect that our research and development expenses will increase due to the continued development of the OPMD program. It is also likely that there will be an increase in the general and administrative expenses due to the obligations of being a domestic public company in the United States.
We had no borrowings as of September 30, 2024 and do not currently have a credit facility.
As of September 30, 2024, we had cash and cash equivalents of approximately $67.8 million. Cash in excess of immediate requirements is invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation. Currently, our cash and cash equivalents are held in bank accounts. . On October 11, 2024, we entered into the Sales Agreement as discussed above, which provides for the sale of up to $75 million of our common stock from time-to-time in “at-the-market offerings”.
The following table sets forth a summary of the net cash flow activity for each of the periods set forth below:
| Three Months Ended | ||||||||
| September 30, | ||||||||
| 2024 | 2023 | |||||||
| (US$’000) | ||||||||
| Net cash provided by (used in): |
||||||||
| Operating activities |
$ | (4,586 | ) | $ | (4,577 | ) | ||
| Investing activities |
— | — | ||||||
| Financing activities |
21,655 | 27,919 | ||||||
| Effects of exchange rate changes on cash and cash equivalents |
(94 | ) | 45 | |||||
|
|
|
|
|
|||||
| Net increase in cash, cash equivalents, and restricted cash |
$ | 67,905 | $ | 25,877 | ||||
|
|
|
|
|
|||||
Operating activities
Net cash used in operating activities for the three months ended September 30, 2024 and 2023 was $4.6 million and $4.6 million, respectively. Net cash used in operating activities was primarily the result of our net loss, partially offset by non-cash expenses, and changes in working capital, including a decrease in payables, trade and other receivables, and prepaid expenses.
47
Table of Contents
Investing activities
Net cash used in investing activities for the three month periods ended September 30, 2024 and 2023 was zero, respectively.
Financing activities
Net cash provided by financing activities was $21.7 million and $27.9 million for the three months ended September 30, 2024 and 2023, respectively. Cash from financing activities in the three months ended September 30, 2024 was related to the issuance of common stock for the exercise of pre-funded warrants, Series 2 warrants, and common warrants, with gross proceeds of $21.7 million. Cash from financing activities in the three months ended September 30, 2023 was related to the issuance of common stock, pre-funded warrants, and common warrants, with gross proceeds of $30.9 million, partially offset by $3.0 million in share issuance costs.
The future of the Company as an operating business will depend on its ability to manage operating costs and budgeted amounts and obtain adequate financing. While we continue to progress discussions and advance opportunities to engage with pharmaceutical companies and continue to seek licensing partners for ddRNAi in disease areas that are not our focus, there can be no assurance as to whether we will enter into such arrangements or what the terms of any such arrangement could be.
While we have established some licensing arrangements, we do not have any products approved for sale and have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize one of our current or future product candidates.
Unless and until we establish significant revenues from licensing programs, strategic alliances or collaboration arrangements with pharmaceutical companies, or from product sales, we anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of product candidates and begin to prepare to commercialize any product that receives regulatory approval. We are subject to the risks inherent in the development of new gene therapy products, and we may encounter unforeseen expenses, difficulties, complications, delays, and other unknown factors that may adversely affect our business. We estimate that our cash and cash equivalents will be sufficient to fund the Company’s operations for at least the next twelve months from the date of this report.
We have based our projections of operating capital requirements on assumptions that may prove to be incorrect and we may use all of our available capital resources sooner than we expect. Because of the numerous risks and uncertainties associated with research, development, and commercialization of pharmaceutical products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| • | the timing and costs of our clinical trials for our ddRNAi and silence and replace product candidates; |
| • | the timing and costs of our preclinical studies for our ddRNAi and silence and replace product candidates; |
| • | the number and characteristics of product candidates that we pursue; |
| • | the outcome, timing, and costs of seeking regulatory approvals; |
| • | revenue received from commercial sales of any of our product candidates that may receive regulatory approval; |
| • | the terms and timing of any future collaborations, licensing, consulting, or other arrangements that we may establish; |
| • | the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights and defending against intellectual property related claims; and |
| • | the extent to which we need to in-license or acquire other products and technologies. |
Contractual Obligations and Commercial Commitments
On October 1, 2016, the Company entered into an operating lease for office space in Hayward, California that originally expired in April 2018. The Company has entered into lease amendments that extended the lease through June 2025. See Note 9 of the Notes to Consolidated Financial Statements included in this Quarterly Report on Form 10-Q.
The Company enters into contracts in the normal course of business with third-party contract research organizations, contract development and manufacturing organizations and other service providers and vendors. These contracts generally provide for termination on notice and, therefore, are cancellable contracts and not considered contractual obligations and commitments.
48
Table of Contents
Critical Accounting Policies and Significant Accounting Estimates
The preparation of consolidated financial statements and related disclosures in conformity with accounting principles generally accepted in the United States of America requires management to make judgments, assumptions and estimates that affect the amounts reported. Note 2 of the Notes to Consolidated Financial Statements included in this Quarterly Report on Form 10-Q describes the significant accounting policies used in the preparation of the consolidated financial statements. Certain of these significant accounting policies are considered to be critical accounting policies.
49
Table of Contents
A critical accounting policy is defined as one that is both material to the presentation of the Company’s consolidated financial statements and requires management to make difficult, subjective, or complex judgments that could have a material effect on the Company’s financial condition or results of operations. Specifically, these policies have the following attributes: (1) the Company is required to make assumptions about matters that are highly uncertain at the time of the estimate; and (2) different estimates the Company could reasonably have used, or changes in the estimate that are reasonably likely to occur, would have a material effect on the Company’s financial condition or results of operations.
Estimates and assumptions about future events and their effects cannot be determined with certainty. The Company bases its estimates on historical experience and on various other assumptions believed to be applicable and reasonable under the circumstances. These estimates may change as new events occur, as additional information is obtained and as the Company’s operating environment changes. These changes have historically been minor and have been included in the consolidated financial statements as soon as they became known. In addition, management is periodically faced with uncertainties, the outcomes of which are not within its control and will not be known for prolonged periods of time. These uncertainties are discussed in the section above entitled “Risk Factors.” Based on a critical assessment of its accounting policies and the underlying judgments and uncertainties affecting the application of those policies, management believes that the Company’s consolidated financial statements are fairly stated in accordance with accounting principles generally accepted in the United States of America and provide a meaningful presentation of the Company’s financial condition and results of operations.
Management believes that the following are critical accounting policies:
Research and Development Expense
Research and development expenses relate primarily to the cost of conducting clinical and preclinical trials. Preclinical and clinical development costs are a significant component of research and development expenses. The Company records accrued liabilities for estimated costs of research and development activities conducted by third-party service providers, which include the conduct of preclinical studies and clinical trials, and contract manufacturing activities. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in trade and other payables on the consolidated balance sheets and within research and development expenses on the consolidated statements of operations and comprehensive loss.
The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers. The Company makes significant judgments and estimates in determining the accrued liabilities balance at the end of each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred.
Share-based Compensation Expense
The Company records share-based compensation in accordance with ASC 718, Stock Compensation. ASC 718 requires the fair value of all share-based employee compensation awarded to employees and non-employees to be recorded as an expense over the shorter of the service period or the vesting period. The Company determines employee and non-employee share-based compensation based on grant-date fair value using the Black-Scholes Option Pricing Model.
Recent Accounting Pronouncements
Accounting Standards recently adopted
ASU 2016-13 – In June 2016, the FASB issued ASU No. 2016-13: “Financial Instruments-Credit Losses (Topic 326)”. This ASU represents a significant change in the accounting for credit losses model by requiring immediate recognition of management’s estimates of current expected credit losses (CECL). Under the prior model, losses were recognized only as they were incurred. The Company adopted this ASU effective July 1, 2023 and determined that its impact on the accompanying consolidated financial statements is immaterial.
Recently Issued Accounting Standards not yet adopted
In December 2023, the FASB issued ASU No. 2023-09, “Income Taxes (Topic 740) – Improvements to Income Tax Disclosures”, which enhances the transparency, effectiveness and comparability of income tax disclosures by requiring consistent categories and greater disaggregation of information related to income tax rate reconciliations and the jurisdictions in which income taxes are paid. This guidance is effective for annual periods beginning after December 15, 2024 with early adoption permitted. The Company is currently evaluating the impact of the ASU on its income tax disclosures within the consolidated financial statements.
50
Table of Contents
In November 2023, the FASB issued ASU No. 2023-07, “Segment Reporting (Topic 280) – Improvements to Reportable Segment Disclosures”, which improves reportable segment disclosure requirements, primarily through enhanced disclosures about significant segment expenses. This ASU also expands disclosure requirements to enable users of financial statements to better understand the entity’s measurement and assessment of segment performance and resource allocation. This guidance is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024, with early adoption permitted. The Company is currently evaluating the impact of the ASU on its disclosures within the consolidated financial statements.
51
Table of Contents
Item 3. Quantitative and Qualitative Disclosures About Market Risk
As a smaller reporting company, we are not required to provide the information pursuant to this Item.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We have established disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended). As of the end of the period covered by this Report we carried out an evaluation under the supervision and with the participation of our management, including our principal executive officer and principal financial and accounting officer, of the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 of the Securities and Exchange Act of 1934, as amended. Based on this evaluation, and as a result of the material weakness in our internal control over financial reporting further described in Management’s Report on Internal Control Over Financial Reporting in Item 9A of our Form 10-K for the fiscal year ended June 30 2024 (relating to our accounting personnel not being able to process and account for complex, non-routine transactions), in accordance with US GAAP due to the Company lacking sufficient personnel and outside consultants with technical accounting expertise to process and account for complex and non-routine transactions), our principal executive officer and principal financial officer, concluded that as of September 30, 2024, our disclosure controls and procedures were not effective to provide reasonable assurance that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer as appropriate to allow timely decisions regarding required disclosure. In order to remediate this matter, we plan to retain the assistance of additional accounting expert to assist in the accounting and reporting of complex, non-routine transactions. We will consider the material weakness to be fully remediated once the applicable controls operate for a sufficient period of time and our management has concluded, through testing, that these controls are operating effectively.
We do not expect that our disclosure controls and procedures or our internal controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Changes in Internal Control over Financial Reporting
Other than the efforts towards remediating the material weakness as previously described above, there were no changes in our internal controls over financial reporting during the quarter ended September 30, 2024 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
52
Table of Contents
Table of Contents
Item 6. Exhibits.
| * | Filed herewith. |
| ** | Furnished, not filed. |
54
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on our behalf by the undersigned thereunto duly authorized.
| Benitec Biopharma Inc. | ||||
| Dated: November 14, 2024 | /s/ Jerel Banks |
|||
| Jerel Banks | ||||
| Executive Chairman and Chief Executive Officer | ||||
| (principal executive officer) | ||||
| /s/ Megan Boston |
||||
| Megan Boston | ||||
| Executive Director (principal financial and accounting officer) | ||||
55